Noncoding DNA
In genetics, noncoding DNA describes components of an organism's DNA sequences that do not encode for protein sequences. In many eukaryotes, a large percentage of an organism's total genome size is noncoding DNA, although the amount of noncoding DNA, and the proportion of coding versus noncoding DNA varies greatly between species.
Much of this DNA has no known biological function and is sometimes referred to as "junk DNA". However, many types of noncoding DNA sequences do have known biological functions, including the transcriptional and translational regulation of protein-coding sequences. Other noncoding sequences have likely, but as-yet undetermined, functions (this is inferred from high levels of homology and conservation seen in sequences that do not encode proteins but, nonetheless, appear to be under heavy selective pressure). While this indicates that noncoding DNA should not be indiscriminately referred to as junk DNA, the lack of function and sequence conservation in a majority of noncoding DNA indicates that much of it may indeed be without function.
Contents |
Fraction of noncoding genomic DNA
The amount of total genomic DNA varies widely between organisms, and the proportion of coding and noncoding DNA within these genomes varies greatly as well. More than 98% of the human genome does not encode protein sequences, including most sequences within introns and most intergenic DNA.[1]
While overall genome size, and by extension the amount of noncoding DNA, are correlated to organism complexity, there are many exceptions. For example, the genome of the unicellular Polychaos dubium (formerly known as Amoeba dubia) has been reported to contain more than 200 times the amount of DNA in humans.[2] The pufferfish Takifugu rubripes genome is only about one eighth the size of the human genome, yet seems to have a comparable number of genes; approximately 90% of the Takifugu genome is noncoding DNA[1] and most of the genome size difference appears to lie in the noncoding DNA. The extensive variation in nuclear genome size among eukaryotic species is known as the C-value enigma or C-value paradox.[3]
About 80 percent of the nucleotide bases in the human genome may be transcribed,[4] but transcription does not necessarily imply function.[5]
Types of noncoding DNA sequences
Noncoding functional RNA
Noncoding RNAs are functional RNA molecules that are not translated into protein. Examples of noncoding RNA include ribosomal RNA, transfer RNA, Piwi-interacting RNA and microRNA.
MicroRNAs are predicted to control the translational activity of approximately 30% of all protein-coding genes in mammals and may be vital components in the progression or treatment of various diseases including cancer, cardiovascular disease, and the immune system response to infection.[6]
Cis- and Trans-regulatory elements
Cis-regulatory elements are sequences that control the transcription of a nearby gene. Cis-elements may be located in 5' or 3' untranslated regions or within introns. Trans-regulatory elements control the transcription of a distant gene.
Promoters facilitate the transcription of a particular gene and are typically upstream of the coding region. Enhancer sequences may also exert very distant effects on the transcription levels of genes.[7]
Introns
Introns are non-coding sections of a gene, transcribed into the precursor mRNA sequence, but ultimately removed by RNA splicing during the processing to mature messenger RNA. Many introns appear to be mobile genetic elements.[8]
Studies of group I introns from Tetrahymena indicate that some introns appear to be selfish genetic elements, neutral to the host because they remove themselves from flanking exons during RNA processing and do not produce an expression bias between alleles with and without the intron.[8] Some introns appear to have significant biological function, possibly through ribozyme functionality that may regulate tRNA and rRNA activity as well as protein-coding gene expression, evident in hosts that have become dependent on such introns over long periods of time; for example, the trnL-intron is found in all green plants and appears to have been vertically inherited for several billions of years, including more than a billion years within chloroplasts and an additional 2–3 billion years prior in the cyanobacterial ancestors of chloroplasts.[8]
Pseudogenes
Pseudogenes are DNA sequences, related to known genes, that have lost their protein-coding ability or are otherwise no longer expressed in the cell. Pseudogenes arise from retrotransposition or genomic duplication of functional genes, and become "genomic fossils" that are nonfunctional due to mutations that prevent the transcription of the gene, such as within the gene promoter region, or fatally alter the translation of the gene, such as premature stop codons or frameshifts.[9] Pseudogenes resulting from the retrotransposition of an RNA intermediate are known as processed pseudogenes; pseudogenes that arise from the genomic remains of duplicated genes or residues of inactivated genes are nonprocessed pseudogenes.[9]
While Dollo's Law suggests that the loss of function in pseudogenes is likely permanent, silenced genes may actually retain function for several million years and can be "reactivated" into protein-coding sequences[10] and a substantial number of pseudogenes are actively transcribed.[9] Because pseudogenes are presumed to change without evolutionary constraint, they can serve as a useful model of the type and frequencies of various spontaneous genetic mutations.[11]
Repeat sequences, transposons and viral elements
Transposons and retrotransposons are mobile genetic elements. Retrotransposon repeated sequences, which include long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs), account for a large proportion of the genomic sequences in many species. Alu sequences, classified as a short interspersed nuclear element, are the most abundant mobile elements in the human genome. Some examples have been found of SINEs exerting transcriptional control of some protein-encoding genes.[12][13][14]
Endogenous retrovirus sequences are the product of reverse transcription of retrovirus genomes into the genomes of germ cells. Mutation within these retro-transcribed sequences can inactivate the viral genome.
Over 8% of the human genome is made up of (mostly decayed) endogenous retrovirus sequences, as part of the over 42% fraction that is recognizably derived of retrotransposons, while another 3% can be identified to be the remains of DNA transposons. Much of the remaining half of the genome that is currently without an explained origin is expected to have found its origin in transposable elements that were active so long ago (> 200 million years) that random mutations have rendered them unrecognizable.[15] Genome size variation in at least two kinds of plants is mostly the result of retrotransposon sequences.[16]
Telomeres
Telomeres are regions of repetitive DNA at the end of a chromosome, which provide protection from chromosomal deterioration during DNA replication.
Functions of noncoding DNA
Many noncoding DNA sequences have important biological functions as indicated by comparative genomics studies that report some regions of noncoding DNA that are highly conserved, sometimes on time-scales representing hundreds of millions of years, implying that these noncoding regions are under strong evolutionary pressure and positive selection.[17] For example, in the genomes of humans and mice, which diverged from a common ancestor 65–75 million years ago, protein-coding DNA sequences account for only about 20% of conserved DNA, with the remaining 80% of conserved DNA is represented in noncoding regions.[18] Linkage mapping often identifies chromosomal regions associated with a disease with no evidence of functional coding variants of genes within the region, suggesting that disease-causing genetic variants lie in the noncoding DNA.[18]
Some specific sequences of noncoding DNA may be features essential to chromosome structure, centromere function and homolog recognition in meiosis.[19]
According to a comparative study of over 300 prokaryotic and over 30 eukaryotic genomes,[20] eukaryotes appear to require a minimum amount of non-coding DNA. This minimum amount can be predicted using a growth model for regulatory genetic networks, implying that it is required for regulatory purposes. In humans the predicted minimum is about 5% of the total genome.
Genetic switches
Some noncoding DNA sequences are genetic "switches" that regulate when and where genes are expressed.[21]
Regulation of gene expression
Some noncoding DNA sequences determine the expression levels of various genes.[22]
Transcription factors
Some noncoding DNA sequences determine where transcription factors attach.[22] A transcription factor is a protein that binds to specific non-coding DNA sequences, thereby controlling the flow (or transcription) of genetic information from DNA to mRNA. Transcription factors act at very different locations on the genomes of different people.
Operators
An operator is a segment of DNA to which a repressor binds. A repressor is a DNA-binding protein that regulates the expression of one or more genes by binding to the operator and blocking the attachment of RNA polymerase to the promoter, thus preventing transcription of the genes. This blocking of expression is called repression.
Enhancers
An enhancer is a short region of DNA that can be bound with proteins (trans-acting factors), much like a set of transcription factors, to enhance transcription levels of genes in a gene cluster.
Promoters
A promoter is a region of DNA that facilitates transcription of a particular gene. Promoters are typically located near the genes they regulate.
Noncoding DNA and evolution
Shared sequences of apparently non-functional DNA are a major line of evidence of common descent.[23]
Pseudogene sequences appear to accumulate mutations more rapidly than coding sequences due to a loss of selective pressure.[11] This allows for the creation of mutant alleles that incorporate new functions that may be favored by natural selection; thus, pseudogenes can serve as raw material for evolution and can be considered "protogenes".[24]
Junk DNA
Junk DNA is a term that was introduced in 1972 by Susumu Ohno,[25] who noted that the mutational load from deleterious mutations placed an upper limit on the number of functional loci that could be expected given a typical mutation rate. Ohno predicted that mammal genomes could not have more than 30,000 loci under selection before the "cost" from the mutational load would cause an inescapable decline in fitness, and eventually extinction. This prediction remains robust, with the human genome containing approximately 20,000 genes. Junk DNA remains a label for the portions of a genome sequence for which no discernible function had been identified. According to a 1980 review in Nature by Leslie Orgel and Francis Crick, junk DNA has "little specificity and conveys little or no selective advantage to the organism".[26] The term is used mainly in popular science and in a colloquial way in scientific publications, and its connotations may have slowed research into the biological functions of noncoding DNA.[27] Several lines of evidence indicate that some "junk DNA" sequences are likely to have unidentified functional activity, and other sequences may have had functions in the past.[28]
Still, a significant amount of the sequence of the genomes of eukaryotic organisms currently appears to fall under no existing classification other than "junk". For example, one experiment removed 0.1% of the mouse genome with no detectable effect on the phenotype.[29] This result suggests that the removed DNA was largely nonfunctional. In addition, these sequences are enriched for the heterochromatic histone modification H3K9me3.[30]
Noncoding DNA and Long range correlations
A statistical distinction between coding and noncoding DNA sequences have been found. It has been observed that nucleotides in non-coding DNA sequences display long range power law correlations while coding sequences do not[31][32][33].
See also
- Conserved non-coding sequence
- Eukaryotic chromosome fine structure
- Phylogenetic footprinting
- Transcriptome
- Intergenic region
- Gene regulatory network
References
- ^ a b Elgar G, Vavouri T (July 2008). "Tuning in to the signals: noncoding sequence conservation in vertebrate genomes". Trends Genet. 24 (7): 344–52. doi:10.1016/j.tig.2008.04.005. PMID 18514361.
- ^ Gregory TR, Hebert PD (April 1999). "The modulation of DNA content: proximate causes and ultimate consequences". Genome Res. 9 (4): 317–24. doi:10.1101/gr.9.4.317. PMID 10207154. http://www.genome.org/cgi/pmidlookup?view=long&pmid=10207154.
- ^ Wahls, W.P., et al. (1990). "Hypervariable minisatellite DNA is a hotspot for homologous recombination in human cells". Cell 60 (1): 95–103. doi:10.1016/0092-8674(90)90719-U. PMID 2295091.
- ^ Pennisi, Elizabeth (2007). "DNA Study Forces Rethink of What It Means to Be a Gene". Science 316 (5831): 1556–7. doi:10.1126/science.316.5831.1556. PMID 17569836.
- ^ Struhl, Kevin (2007). "Transcriptional noise and the fidelity of initiation by RNA polymerase II". Nature Structural & Molecular Biology 14 (2): 103–105. doi:10.1038/nsmb0207-103. PMID 17277804.
- ^ Li M, Marin-Muller C, Bharadwaj U, Chow KH, Yao Q, Chen C (April 2009). "MicroRNAs: Control and Loss of Control in Human Physiology and Disease". World J Surg 33 (4): 667–84. doi:10.1007/s00268-008-9836-x. PMC 2933043. PMID 19030926. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2933043.
- ^ Visel A, Rubin EM, Pennacchio LA (September 2009). "Genomic Views of Distant-Acting Enhancers". Nature 461 (7261): 199–205. Bibcode 2009Natur.461..199V. doi:10.1038/nature08451. PMC 2923221. PMID 19741700. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2923221.
- ^ a b c Nielsen H, Johansen SD (2009). "Group I introns: Moving in new directions". RNA Biol 6 (4): 375–83. doi:10.4161/rna.6.4.9334. PMID 19667762. http://www.landesbioscience.com/journals/rna/abstract.php?id=9334.
- ^ a b c Zheng D, Frankish A, Baertsch R, et al. (June 2007). "Pseudogenes in the ENCODE regions: Consensus annotation, analysis of transcription, and evolution". Genome Res. 17 (6): 839–51. doi:10.1101/gr.5586307. PMC 1891343. PMID 17568002. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1891343.
- ^ Marshall CR, Raff EC, Raff RA (December 1994). "Dollo's law and the death and resurrection of genes". Proc. Natl. Acad. Sci. U.S.A. 91 (25): 12283–7. Bibcode 1994PNAS...9112283M. doi:10.1073/pnas.91.25.12283. PMC 45421. PMID 7991619. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=45421.
- ^ a b Petrov DA, Hartl DL (2000). "Pseudogene evolution and natural selection for a compact genome". J. Hered. 91 (3): 221–7. doi:10.1093/jhered/91.3.221. PMID 10833048. http://jhered.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=10833048.
- ^ Ponicsan SL, Kugel JF, Goodrich JA (February 2010). "Genomic gems: SINE RNAs regulate mRNA production". Curr Opin Genet Dev 20 (2): 149–55. doi:10.1016/j.gde.2010.01.004. PMC 2859989. PMID 20176473. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2859989.
- ^ Häsler J, Samuelsson T, Strub K (July 2007). "Useful 'junk': Alu RNAs in the human transcriptome". Cell. Mol. Life Sci. 64 (14): 1793–800. doi:10.1007/s00018-007-7084-0. PMID 17514354.
- ^ Walters RD, Kugel JF, Goodrich JA (Aug 2009). "InvAluable junk: the cellular impact and function of Alu and B2 RNAs". IUBMB Life 61 (8): 831–7. doi:10.1002/iub.227. PMID 19621349.
- ^ International Human Genome Sequencing Consortium (February 2001). "Initial sequencing and analysis of the human genome". Nature 409 (6822): 879–888. doi:10.1038/35057062. PMID 11237011.
- ^ [1] [2]
- ^ Ludwig MZ (December 2002). "Functional evolution of noncoding DNA". Curr. Opin. Genet. Dev. 12 (6): 634–9. doi:10.1016/S0959-437X(02)00355-6. PMID 12433575. http://linkinghub.elsevier.com/retrieve/pii/S0959437X02003556.
- ^ a b Cobb J, Büsst C, Petrou S, Harrap S, Ellis J (April 2008). "Searching for functional genetic variants in non-coding DNA". Clin. Exp. Pharmacol. Physiol. 35 (4): 372–5. doi:10.1111/j.1440-1681.2008.04880.x. PMID 18307723.
- ^ Subirana JA, Messeguer X (March 2010). "The most frequent short sequences in non-coding DNA". Nucleic Acids Res. 38 (4): 1172–81. doi:10.1093/nar/gkp1094. PMC 2831315. PMID 19966278. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2831315.
- ^ S. E. Ahnert, T. M. A. Fink and A. Zinovyev (2008). "How much non-coding DNA do eukaryotes require?" (PDF). J Theor. Biol. 252 (4): 587–592. doi:10.1016/j.jtbi.2008.02.005. PMID 18384817. http://www.tcm.phy.cam.ac.uk/~tmf20/PUBLICATIONS/jtb_07.pdf.
- ^ Carroll, Sean B., et al. (May 2008). "Regulating Evolution". Scientific American 298 (5): 60–67. doi:10.1038/scientificamerican0508-60. PMID 18444326.
- ^ a b Callaway, Ewen (March 2010). "Junk DNA gets credit for making us who we are". New Scientist. http://www.newscientist.com/article/dn18680-junk-dna-gets-credit-for-making-us-who-we-are.html?full=true&print=true.
- ^ "Plagiarized Errors and Molecular Genetics", talkorigins, by Edward E. Max, M.D., Ph.D.
- ^ Balakirev ES, Ayala FJ (2003). "Pseudogenes: are they "junk" or functional DNA?". Annu. Rev. Genet. 37: 123–51. doi:10.1146/annurev.genet.37.040103.103949. PMID 14616058.
- ^ So much "junk" DNA in our genome, In Evolution of Genetic Systems (1972). H. H. Smith. ed. Gordon and Breach, New York. pp. 366–370.
- ^ Orgel LE, Crick FH (April 1980). "Selfish DNA: the ultimate parasite". Nature 284 (5757): 604–7. Bibcode 1980Natur.284..604O. doi:10.1038/284604a0. PMID 7366731. http://www.nature.com/nature/journal/v284/n5757/abs/284604a0.html.
- ^ Khajavinia A, Makalowski W (May 2007). "What is "junk" DNA, and what is it worth?". Scientific American 296 (5): 104. doi:10.1038/scientificamerican0307-104. PMID 17503549. http://www.scientificamerican.com/article.cfm?id=what-is-junk-dna-and-what. "The term "junk DNA" repelled mainstream researchers from studying noncoding genetic material for many years"
- ^ Biémont, Christian; Vieira, C (2006). "Genetics: Junk DNA as an evolutionary force". Nature 443 (7111): 521–4. Bibcode 2006Natur.443..521B. doi:10.1038/443521a. PMID 17024082.
- ^ M.A. Nobrega, Y. Zhu, I. Plajzer-Frick, V. Afzal and E.M. Rubin (2004). "Megabase deletions of gene deserts result in viable mice". Nature 431 (7011): 988–993. Bibcode 2004Natur.431..988N. doi:10.1038/nature03022. PMID 15496924.
- ^ Rosenfeld, Jeffrey A; Wang, Zhibin; Schones, Dustin; Zhao, Keji; DeSalle, Rob; Zhang, Michael Q (31 March 2009). "Determination of enriched histone modifications in non-genic portions of the human genome". BMC Genomics 10: 143. doi:10.1186/1471-2164-10-143. PMC 2667539. PMID 19335899. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2667539.
- ^ C.-K. Peng, S. V. Buldyrev, A. L. Goldberger, S. Havlin, F. Sciortino, M. Simons, H. E. Stanley; Buldyrev, SV; Goldberger, AL; Havlin, S; Sciortino, F; Simons, M; Stanley, HE (1992). "Long-range correlations in nucleotide sequences". Nature 356 (6365): 168–70. doi:10.1038/356168a0. PMID 1301010. http://havlin.biu.ac.il/Publications.php?keyword=Long-range+correlations+in+nucleotide+sequences&year=*&match=all.
- ^ W. Li and, K. Kaneko (1992). "Long-Range Correlation and Partial
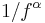 Spectrum in a Non-Coding DNA Sequence". Europhys. Lett 17: 655–660. http://chaos.c.u-tokyo.ac.jp/papers/bio0/wli.pdf.
Spectrum in a Non-Coding DNA Sequence". Europhys. Lett 17: 655–660. http://chaos.c.u-tokyo.ac.jp/papers/bio0/wli.pdf. - ^ S. V. Buldyrev, A. L. Goldberger, S. Havlin, R. N. Mantegna, M. Matsa, C.-K. Peng, M. Simons, and H. E. Stanley; Goldberger, A.; Havlin, S.; Mantegna, R.; Matsa, M.; Peng, C.-K.; Simons, M.; Stanley, H. (1995). "Long-range correlations properties of coding and noncoding DNA sequences: GenBank analysis". Phys. Rev. E 51 (5): 5084. doi:10.1103/PhysRevE.51.5084. http://havlin.biu.ac.il/Publications.php?keyword=Long-range+correlations+properties+of+coding+and+noncoding+DNA+sequences:+GenBank+analysis&year=*&match=all.
- Bennett, M.D. and I.J. Leitch (2005). "Genome size evolution in plants". In T.R. Gregory (ed.). The Evolution of the Genome. San Diego: Elsevier. pp. 89–162.
- Gregory, T.R (2005). "Genome size evolution in animals". In T.R. Gregory (ed.). The Evolution of the Genome. San Diego: Elsevier. ISBN 0123014638.
- Shabalina SA, Spiridonov NA (2004). "The mammalian transcriptome and the function of non-coding DNA sequences". Genome Biol. 5 (4): 105. doi:10.1186/gb-2004-5-4-105. PMC 395773. PMID 15059247. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=395773.
- Castillo-Davis CI (October 2005). "The evolution of noncoding DNA: how much junk, how much func?". Trends Genet. 21 (10): 533–6. doi:10.1016/j.tig.2005.08.001. PMID 16098630.